Running OpenMMDL Setup
This page displays the preparation paths and forcefields available in OpenMMDL and showcases the application of OpenMMDL Setup.

To start the OpenMMDL Setup we need to activate the openmmdl environment. To do this we have to enter the following command lines:
conda activate openmmdl
Now that we have activated the openmmdl environment we can start OpenMMDL Setup. To do this you need to type the following:
openmmdl setup
This will open the OpenMMDL Setup, which you can use for the creation of the input files for OpenMMDL Simulation.
There are two possible options to create the input files for OpenMMDL Simulation:
1. The PDBFixer path, where a pdb file of the protein is used as an input for the preparation and simulation. The tutorial for the PDBFixer path can be found here.
Here is the table of the currently available forcefields and water models for the PDBFixer path:
Water model |
AMBER14 / AMBER19 |
AMBER99SB / AMBER99SB-ILDN / AMBER03 / AMBER10 |
|---|---|---|
TIP3P |
✓ |
✓ |
TIP3P-FB |
✓ |
✓ |
SPC/E |
✓ |
✓ |
TIP4P-Ew |
✓ |
✓ |
TIP4P-FB |
✓ |
— |
TIP5P |
— |
✓ |
OPC3 |
✓ |
✓ |
OPC |
✓ |
✓ |
Water model |
CHARMM36 |
CHARMM36 2024 |
|---|---|---|
SPC/E |
✓ |
✓ |
TIP4P-Ew |
✓ |
✓ |
TIP5P |
✓ |
✓ |
CHARMM default |
✓ |
✓ |
TIP3P-PME-B |
✓ |
✓ |
TIP3P-PME-F |
✓ |
✓ |
TIP4P-2005 |
✓ |
✓ |
TIP5P-Ew |
✓ |
✓ |
2. The Amber path, where prmtop and inpcrd files are used the preparation and simulation. This path allows us to either use already prepared prmtop and inpcrd as an input or create the prmtop and inpcrd from PDB files of the receptor and ligand. The tutorial for the Amber path can be found here.
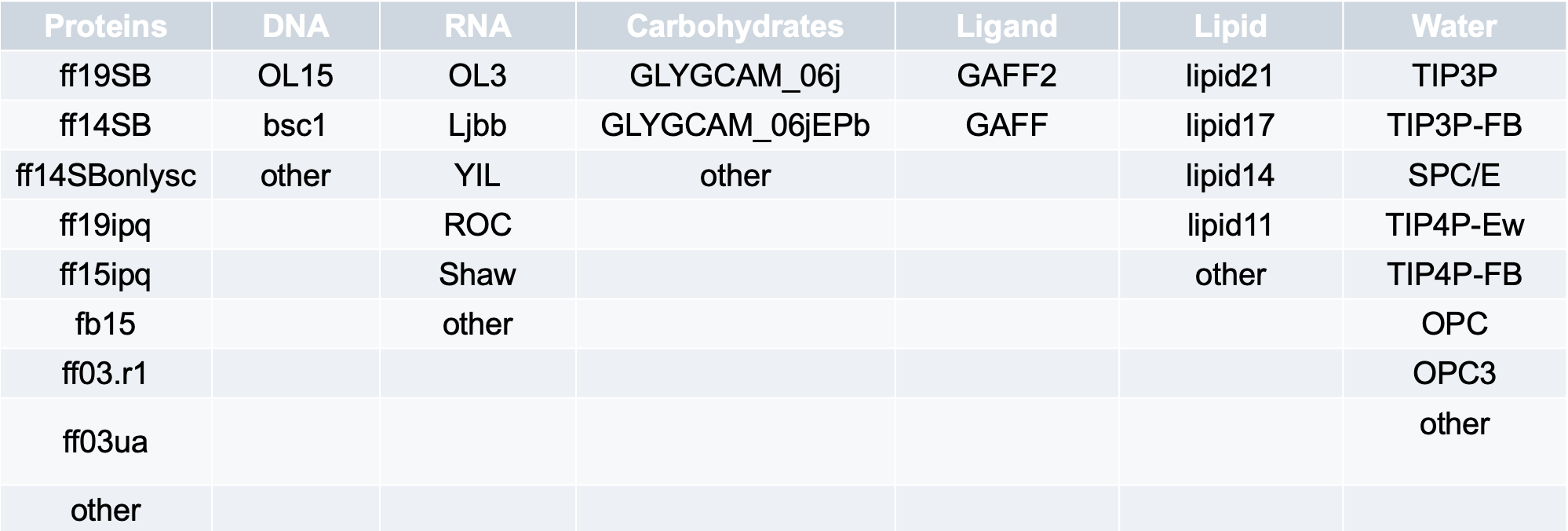
In the table, the first row is the default setting, and the term other allows users to type their desired forcefields from those accessible in AmberTools 22.0 into the designated textbox.