Amber Path
Introduction
OpenMM enables users to perform molecular dynamics (MD) simulations using AMBER topology (.prmtop) and coordinate (.inpcrd) files as input. However, obtaining these essential Amber files can be a daunting task for users unfamiliar with Amber commands and Bash scripting. To address this challenge, OpenMMDL Setup provides a user-friendly interface, streamlining the process of generating the required Amber files for subsequent MD simulations.
In this tutorial, we will use the mu opioid receptor (PDB ID: 8EFO) as an illustrative example to provide a comprehensive, step-by-step guide on configuring and initiating an MD simulation using Amber files prepared by OpenMMDL Setup.
Starting OpenMMDL Setup
We start the tutorial by creating an folder to store input files and facilitate the MD simulation.
Create a new folder we enter the following command lines in the terminal:
mkdir openmmdl_amber_tutorial
Copy the receptor and ligand PDB files from /openmmdl/openmmdl_simulation/tutorial_systems/amber_path/8efo_membrane into the newly created folder:
copy 8EFO_protein.pdb openmmdl_amber_tutorial
copy 8QY.pdb openmmdl_amber_tutorial
In practice, replace the PDB files with the specific files you intend to simulate.
Activate the OpenMMDL environment by entering the following command:
conda activate openmmdl
Launch OpenMMDL Setup:
openmmdl setup
Selecting the Amber Path
The first step is to choose the Amber path by clicking the “Amber” button.

Selecting the input files
The second step involves choosing input files. Two options are available:

1. Yes, my files are already to simulate. When selected, users can choose the Prmtop and Inpcrd files via the file browser. Clicking “Continue” transitions to the ‘Simulation Setup’ page.
If the above system contains ligands and conducting MD post-analysis using OpenMMDL is needed, checking the ligand box based on the ligand type, and then type the resname of the ligand in the prmtop file in the textfield.
Tip
To find the resname of the ligand, open the prmtop file in a text editor and search for ‘%FLAG RESIDUE_LABEL’. The resname of the ligand should be the last one listed after the common amino acids and before the lipids and water molecules. For example, in the image below, the ligand is labeled as ‘UNL’ in this prmtop file.

2. No, I want to prepare them here. When selected, clicking “Continue”, users are directed to the ‘Amber Configuration’ page.
For this tutorial, we will select the second option.
Amber Configuration
This page allows users to configure Amber settings and generate the Bash script for Amber files generation.

The page consists of three tabs: Receptor, Ligand, and Add Water/Membrane.
Receptor
Depending on the macromolecule type (Protein, DNA, RNA, or Carbohydrate), users can select the receptor PDB file using the file browser and choose the appropriate force fields.
If needed, users can specify an ‘Other Force Field’ in the provided textbox. To do this, select ‘other’ from the drop-down menu of “Force Field,” and the ‘Other Force Field’ textbox will appear. Users can input the force field name in the textbox.
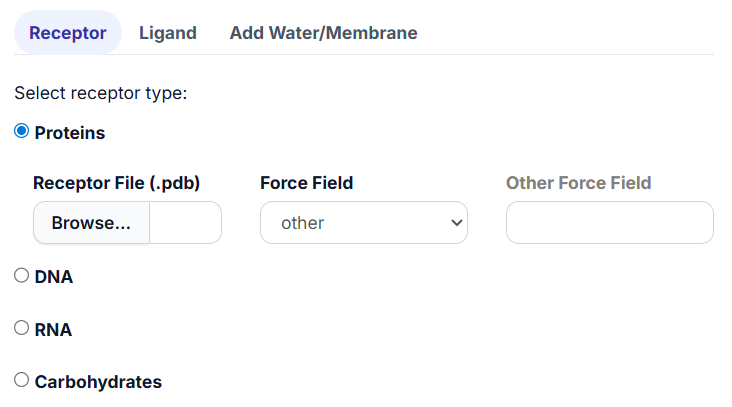
Note: Only force fields provided by AmberTools are supported. To check available force fields, navigate to /home/user/miniconda3/envs/openmmdl/dat/leap/cmd. Replace the path with the location of your OpenMMDL installation.
Users can select one type of macromolecule from the options listed above at a time.
In this tutorial, we will select the protein option, navigate to the folder ‘openmmdl_amber_tutorial’, and select’8EFO_protein.pdb’,and select ‘ff19SB’ as the receptor force field.
Ligand
Depends on the type of ligand the user intends to simulate, two options are available:
2.1 Normal Ligand.
It refers to the small molecule that is made up of C, N, O, S, P, H, F, Cl, Br and I, thus is compatible with GAFF and can be processed by Antechamber and Parmchk2.
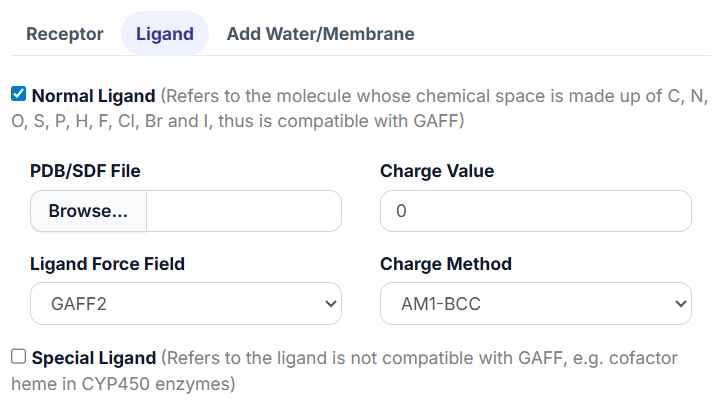
Upon selecting the ‘Normal Ligand’ option, the parameter settings for the ligand will be revealed.
Begin by clicking the “Browse…” button to select the ligand PDB or SDF file.
Fill in the charge value for the ligand in the ‘Charge Value’ textbox; this value should be an integer (e.g., -1 or 2).
Tip
For PDB file, the charge value can be calculated by opening the ligand PDB file in a text editor and summing up the values in the last column of the file.
For SDF file, the charge value can be calculated by opening the ligand SDF file in a text editor and go to the line where starts with ‘M CHG’. For example, ‘M CHG 3 14 -1 38 -1 40 1’ stands for that totally 3 atoms have charge, add -1 charge to atom 14 and 38, and add 1 charge to atom 40. The charge value is -1+(-1)+1=-1.
Choose the ‘Ligand Force Field’ from the available options: General Amber Force Field (GAFF) or GAFF2.
Finally, select the ‘Charge Method’ from the drop-down menu.
The obabel tool will be utilized to convert the ligand file format between PDB and SDF formats.
Warning: When the file format is pdb, the prefix of the filename should be the same as the ligand name in the PDB file. For instance, the ligand name in the PDB file is ‘8QY’, and the filename should be ‘8QY.pdb’.
In this tutorial, we will select the ligand ‘8QY.pdb’, set the charge value to 1, select the ‘GAFF2’ force field, and choose the ‘bcc’ charge method.
2.2 Special Ligand.
For ligands that Antechamber cannot process, such as cofactors like heme in CYP450 enzymes, users can check the ‘Special Ligand’ option. The AMBER parameter database serves as a valuable source for finding Amber files for these special ligands. Follow the guidance provided in the application to set up the generation of Amber files for the special ligand.
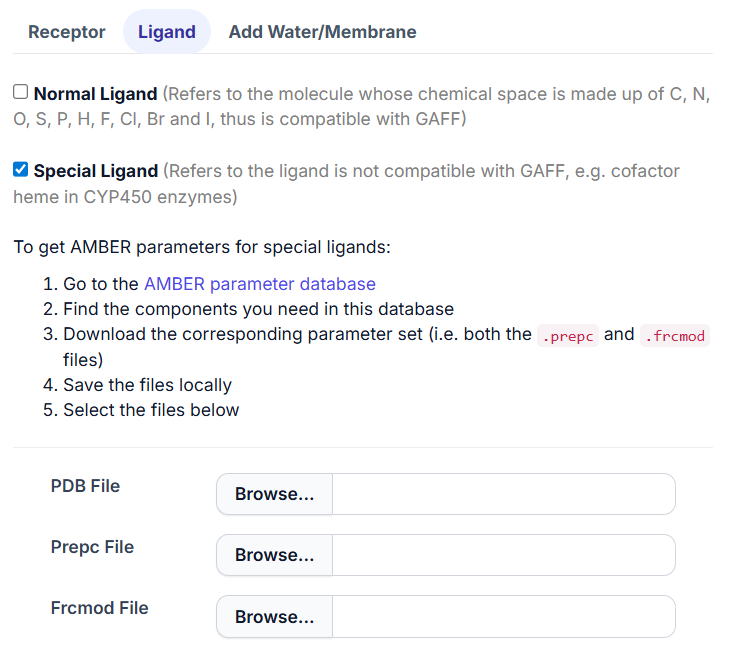
Warning: As the special ligand usually comes with receptor from RCSB website, here we would ask the user to submit it only in pdb format.
Users can select either one or both of the above types of ligands at one time.
Add Water/Membrane
Depending on the environment of the biosystem, users should consider adding water or a membrane. Choose between ‘Add Water Box’ or ‘Add Membrane and Water’ in this tab.
3.1 Add water Box.
When this option is selected, users can further select the ‘Box Type’ from the drop-down list and then specify the ‘Distance (Å)’ value in the textbox.
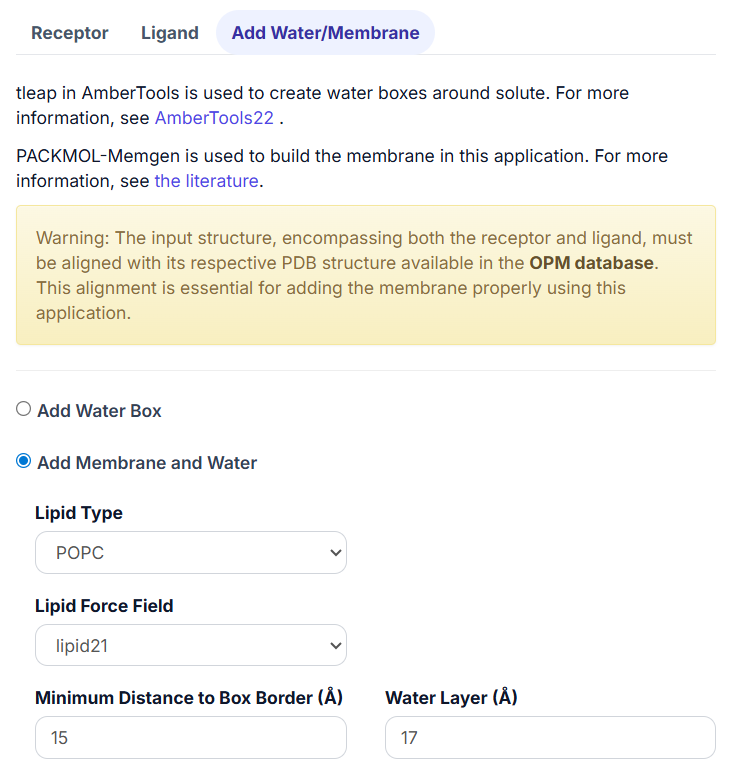
3.2 Add Membrane and Water.
When this option is selected, users can further select the ‘Lipid Type’ and ‘Lipid Force Field’ from the drop-down list.
If the listed lipid type does not match the desired one, click on the ‘Other Type or Mixture’ option. Then, input the lipid type in the pop-up textbox of ‘Other Types or Mixture’ and set the ‘Lipid Ratio’. For instance, ‘POPC:TOPC’ in ‘Other Types or Mixture’ and ‘1:1’ in the ‘Lipid Ratio’ means the membrane consists of 1 POPC and 1 TOPC.
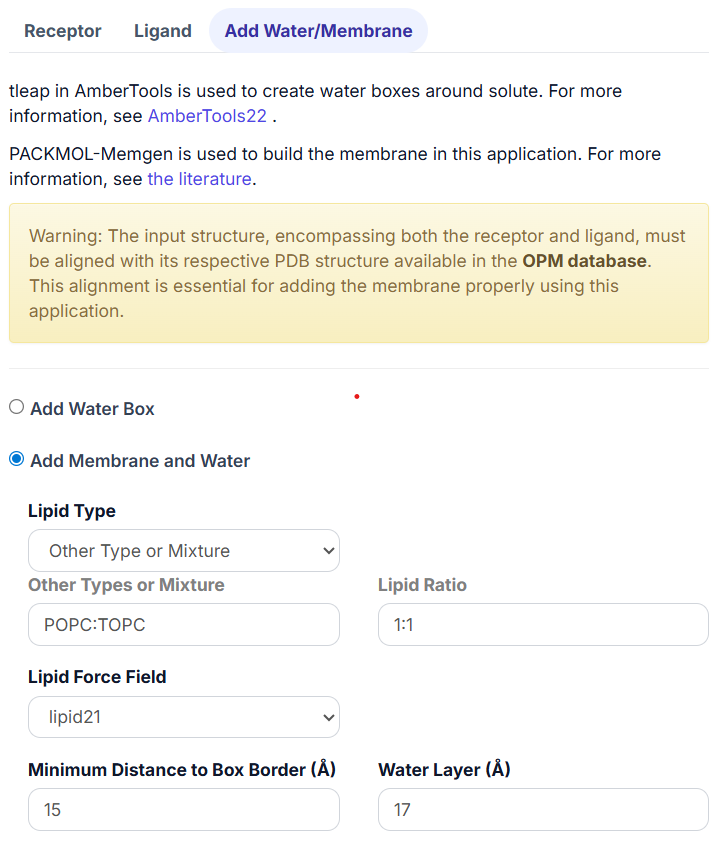
When selecting only one type of lipid, set the ‘Lipid Ratio’ to 1.
Warning: Ensure that the input structure, including both the receptor and ligand, aligns with their respective PDB structures available in the OPM database. Proper alignment is crucial for adding the membrane accurately using this application.
In this tutorial, we will select the ‘add Membrane and Water’ option, and keep the default values for all parameters.
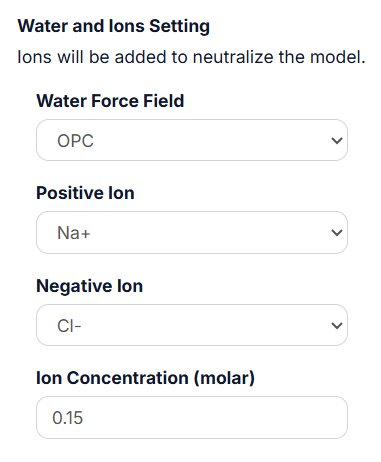
3.3 Water and Ions Setting.
It is a must for both ‘Add water Box’ and ‘Add Membrane and Water’ options. The Ions will be added to neutralize the model. The user can select the ‘Water Force Field’, ‘Positive Ion’ and ‘Negative Ion’ in the drop-down list, and then type the ‘Ion Concentration (molar)’ value in the textbox.
Save Script
Click ‘Save Script’ on the top of the right code block to download the generated Bash script based on the configuration. Save it in the previously created tutorial folder. Click ‘Continue’ to proceed to the ‘Simulation Setup’ page.
Simulation Setup
Configure simulation options across five tabs: System, Integrator, Simulation, Output, and MDAnalysis. Click ‘Save Script’ to download the generated Python script based on the configuration, saving it in the tutorial folder.
Run Bash Script
In the terminal, navigate to the ‘openmmdl_amber_tutorial’ folder and run the Bash script to generate Amber files:
bash run_ambertools.sh
If the script runs not successfully, please check the error message in the output ‘leap.log’ file and modify the input PDB files accordingly.Then go back to the ‘Amber Configuration’ page to regenerate the Bash script and run it again.
Run MD simulation
Once the Prmtop and Inpcrd files are generated, the user can run the MD simulation by typing the following command lines:
python3 OpenMMDL_Simulation.py
Or run the several MD recplicas via slurm.The run_slurm.sh is in tutorial folder /openmmdl/openmmdl-simulation/tuturial_systems/amber_path/8efo_membrane. Firstly copy it to the tutorial folder
copy run_slurm.sh openmmdl_amber_tutorial
Remember to replace the slurm configuration and environment openmmdl path with your own via a text editor. Finally run the following command lines:
sbatch run_slurm.sh